| What is I-TASSER-MTD? |
| How does I-TASSER-MTD generate multi-domain protein structure predictions? |
In the second step, if the query sequence is predicted as a multi-domain protein and none of the top 10 threading templates can cover all domains (i.e., >=1 domains with the alignment coverage smaller than the cutoff C=0.95), each domain model will be independently generated by D-I-TASSER, an new version of I-TASSER improved by integrating deep learning predicted spatial restraints. Otherwise, if the query is predicted as a single-domain protein or >=1 top templates can cover all domains, the structure is directly modelled by D-I-TASSER.
In the third step, the domain models are assembled into the full-length model by DEMO guided by the structurally analogous templates, the inter-domain distances and domain-domain intefaces predicted by DeepPotential, and the knowledge-based inter-domain potentials.
In the last step, the protein function, including the Enzyme Commission (EC) numbers, Gene Ontology (GO) terms, and ligand-binding sites are predicted by COFACTOR for the individual domain and full-chain protein based on the modeled structures, sequences, and protein-protein interactions (PPIs).
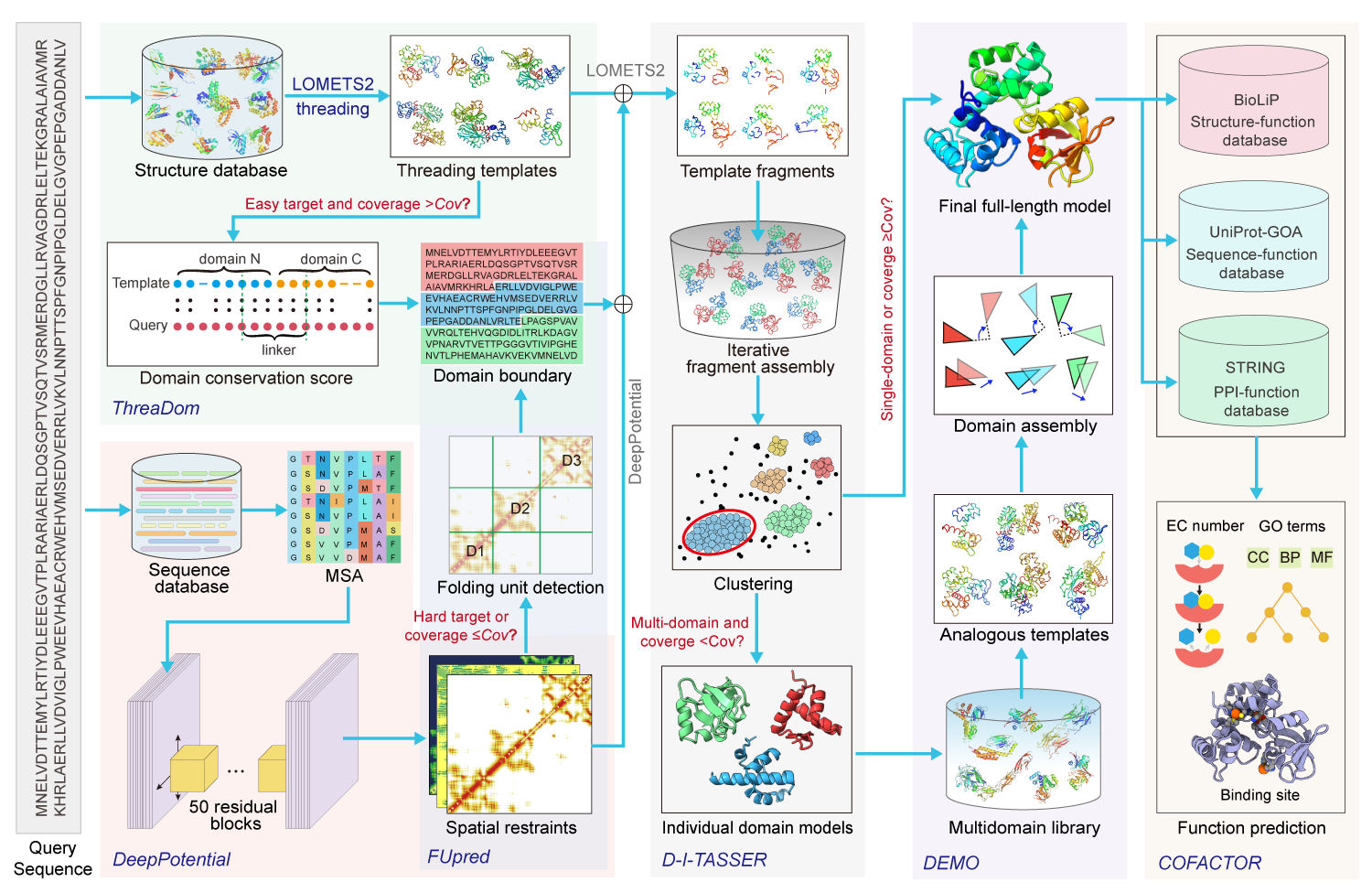
Figure 1. Pipeline of I-TASSER-MTD for automated multi-domain protein structruce and function prediction.
| What are the performances of I-TASSER-MTD server compared with other methods? |
We have used the I-TASSER-MTD protocol (as ‘Zhang-Server’) to predict structures of all targets in the latest CASP13 and CASP14, and the protocol was ranked as the number one server. Figures 2a and 2b show the comparisons between the I-TASSER-MTD protocol and other top 4 servers for protein structure prediction in CASP13 and CASP 14, respectively, where we sorted them according to the GDT-score of full-length models of multi-domain proteins. As shown in the figure, the performance of I-TASSER-MTD on the full-length structure prediction of multi-domain proteins is better than other servers for both CASP13 and CASP14 targets. The accuracy of individual domain models of multi-domain proteins is also better than other severs as each individual domain is independently modelled. In particular, I-TASSER-MTD achieves an average GDT-score of 65.5 for all individual domain models of multi-domain proteins in CASP14, which is 8.3% higher than that of the second best server, Yang-Server (60.5). In addition to multi-domain protein structure prediction, the protocol also achieves a superior performance on single-domain protein structure modeling. Especially in CASP14, the performance of Zhang-Server is clearly better than other third-party servers. Furthermore, I-TASSER-MTD can accurately distinguish multi-domain proteins from single-domain and predict the domain boundary with high accuracy.
The performance of I-TASSER-MTD on the domain boundary prediction is also compared with two state-of-the-art methods ConDO and DoBo over the CASP13 and CASP14 targets. As shown in Figs. 2c and 2d, the performance of I-TASSER-MTD for domain boundary prediction is significantly better than these two methods in terms of normalized domain overlap (NDO) score for the protein domain boundary prediction, and accuracy (ACC) and Matthew’s correlation coefficient (MCC) for the protein classification. The performance of I-TASSER-MTD is obviously improved in CASP14 due to the use of FUpred, which is guided by the deep learning predicted contact map.
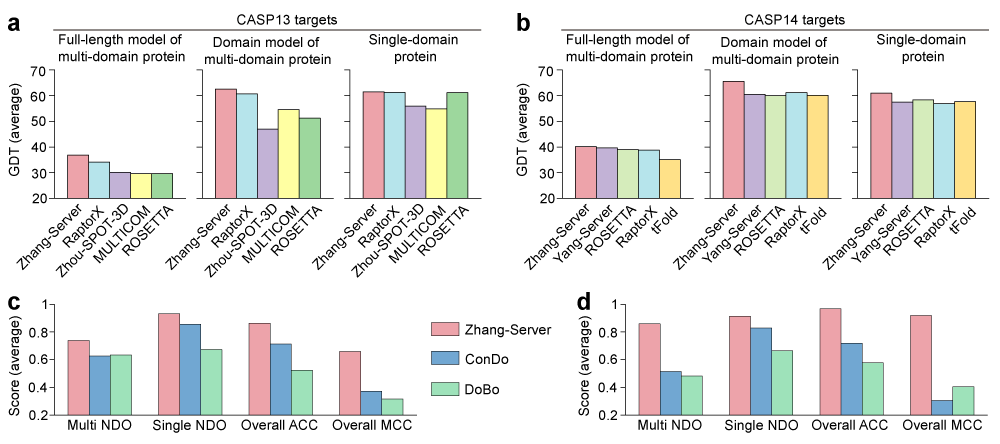
Figure 2. Performance of the I-TASSER-MTD protocol on the CASP13 and CASP14 targets. a and b, Comparison between I-TASSER-MTD (Zhang-Server) with other top 4 servers in CASP13 (a) and CASP14 (b) on the performance of full-length multi-domain proteins, individual domains of multi-domain proteins, and single-domain proteins in terms of the global distance test (GDT) score, where the server are sorted according to the GDT score of full-length models of multi-domain proteins. c and d, Comparison of I-TASSER-MTD with ConDo and DoBo for the protein domain boundary prediction over the CASP13 (c) and CASP14 (d) targets
| What are the output of the I-TASSER-MTD server if you submit a seqeunce? |
| How to interpret the output data generated by the I-TASSER-MTD server? |
For each target, I-TASSER-MTD reports up to five full-length models ranked by the total energy. It is possible that the lower-rank models have a higher eTM-score. Although the first model has a higher eTM-score and a better quality in most cases, it is not unusual that the lower-rank models have a better quality than the higher-rank models. If only one full-length model is reported, it indicates that the model is directly generated by D-I-TASSER as the threading templates can cover all domains, and the top templates identified by LOMETS have consistent topologies. In these cases, the final model usually has a relative high eTM-score, indicating a high-quality final model.
I-TASSER-MTD identifies the analogous full-length templates from a non-redundant multi-domain protein library using TM-align structural alignments. All domain models are aligned to each template of the library by TM-align, and the TM-score of all domains is defined as the score of a template. The top 10 templates with the highest score are selected to generate the initial full-length model and deduce the inter-domain distance restraints to guide the domain assembly.
eTM-score is an estimated TM-score for estimating the quality of predicted models by I-TASSER-MTD. It is calculated based on the convergence parameters of the domain assembly simulations, the quality of the full-length templates for domain assembly, the consistency between the deep learning predicted inter-domain distances/interfaces and that in the assembled model, and the estimated accuracy of the individual domain. eTM-score is typically in the range of [0,1], where a eTM-score of higher value signifies a model with a high confidence and vice-versa.
TM-score is a metric for measuring the structural similarity between two structures (see Zhang and Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins, 2004 57: 702-710). The purpose of proposing TM-score is to solve the problem of RMSD which is sensitive to the local error. Because RMSD is an average distance of all residue pairs in two structures, a local error (e.g. a misorientation of the tail) will arise a big RMSD value although the global topology is correct. In TM-score, however, the small distance is weighted stronger than the big distance which makes the score insensitive to the local modeling error. A TM-score >0.5 indicates a model of correct topology and a TM-score <0.17 means a random similarity. These cutoff does not depends on the protein length.
After the full-length model generation, I-TASSER-MTD uses TM-align program to match the first model to all structures in the PDB library. This section reports the top 10 proteins from the PDB which have the closest structural similarity (i.e. the highest TM-score) to the predicted I-TASSER-MTD model. Due to the structural similarity, these proteins often have similar function to the target. However, users are encouraged to use the predicted function by COFACTOR to infer the biological function of the target protein, since COFACTOR has been extensively trained to derive function from multi-source of sequence and structure features which has on average a much higher accuracy than the function annotations derived only from the global structure comparison.
| How to use known information (e.g. full-length templates, experimental data) to improve I-TASSER-MTD modeling? |
The I-TASSER-MTD server currently accepts the following information:
| How long does it take for I-TASSER-MTD to generate the predictions for your protein? |
However, it will cost less time if you provide the domain definition since the program does not need to predict the domain bounaries.
| How to cite I-TASSER-MTD? |
| Funding support |
| Contact information |
yangzhanglab![]() umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218
umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218