I. Understanding the input.
Mutation format:
SSIPe accepts mutations in the following format, which include 'WT residue', 'Chain ID', 'residue position in chain', 'mutant residue', example:
QA22D;
HA18F,QA22D;
HA18F,MB20A;
Note:
- One mutation set per line and each line ends in semicolon.
- If one mutation set has more than one mutation, separate them by commas.
- In the above example, the first line represents a single mutation
on Chain A where Glu22 is mutated to Asp. The second line represents a double
mutation on the same chain: His18 on chain A is mutated to Phe and Glu22 on chain A is mutated to Asp.
The third line represents a double mutation on different chains: His18 on chain A is mutated to Phe
and Met20 on chain B is mutated to Ala.
II. Understanding the output.
-
Binding affinity change upon mutations
The binding affinity between two proteins is measured by Gibbs
free energy change ΔGbind=Gcomplex-Gmonomers when two monomers form a
complex. The more negative ΔG is, the stronger the binding is. The
effect of mutation on binding affinity is measured by different of free energy
change between mutant and wild type: ΔΔGbind,wt→mut = ΔGbind,mut - ΔGbind,wt.
In SSIPe, a favorable mutation is defined as a mutation that has ΔΔG < -0.5 kcal mol-1, while an unfavorable mutation is defined as a mutation that has ΔΔG > 0.5 kcal mol-1; otherwise neutral (i.e., 0.5 kcal mol-1 ≤ ΔΔG ≤ 0.5 kcal mol-1).
-
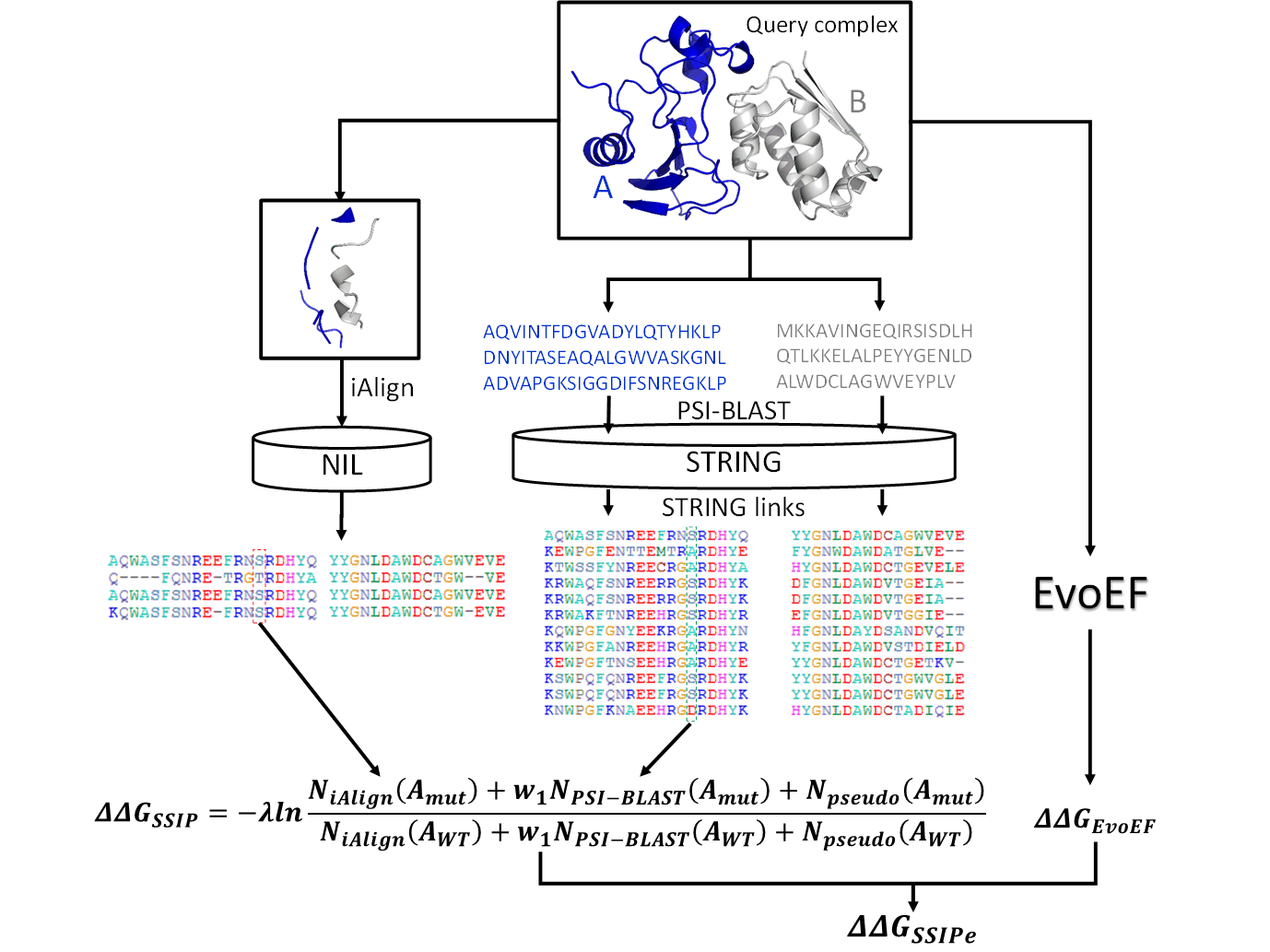
Structure-based interface MSA by iAlign from NIL
SSIPe uses iAlign to do pairwise alignment of the interface structure of query complex to
a set of non-redundant protein-protein interface structures (NIL) built from the PIFACE database.
interface alignment, a pair of interface sequence profile is constructed.
-
Sequence-based interface MSA by PSI-BLAST from STRING
SSIPe also uses PSI-BLAST to do pairwise alignment of the two monomer protein sequences to the STRING
sequence database, and then the interface alignment is extracted from the whole sequence alignment based on protein-protein interface defined by iAlign.
III. Options.
-
Interface similarity cutoff: The default cutoff is IS-score = 0.5.
IS-score cutoff: IS-score should be in [0,1], with a higher value indicating a higher interface structural similarity.
Based on our benchmark test, IS-score cutoff in the range of [0.45, 0.55] gives good prediction performance, and a value of 0.5 gives the best prediction performance.
To avoid using IS-score cutoff incorrectly, the real cutoff for calculation is resticted into [0.45, 0.55] if the value is not in this range. Otherwise, the user-provided
value is used for prediction.
-
Forcefield to use:
The SSIPe server provides three forcefield engines for ΔΔGbind prediction.
SSIPE (default): SSIPe by default combines ΔΔGSSIP derived from structure and sequence alignment and ΔΔGEvoEF calculated by
physics-based energy function EvoEF, which exhibits better ΔΔG prediction than using SSIP or EvoEF only.
SSIP: only use structure and sequence-based interface profiles. This is important when the user knows that a mutation can cause big shift of backbone conformation, where
the physical energy function may result in big prediction errors.
EvoEF: only use physcial energy function EvoEF. Compared with SSIPE and SSIP, EvoEF doesn't search structure and sequence homologs from large databases and has the fastest
speed. In addition, it will generate all the mutant models using the user-provided initial complex structure. User can download the standalone EvoEF program at
https://zhanglab.dcmb.med.umich.edu/EvoEF.
|