Overview
LOMETS (LOcal MEta-Threading-Server, version 3) is a meta-server method for protein structure prediction [1-3] and function annotation. It generates protein structure predictions by ranking and selecting models from multiple state-of-the-art threading programs. Starting from a query sequence, deep multiple sequence alignment (MSA) are generated by iterative sequence homology searches through multiple sequence databases. Then the MSA is fed into DeepPotential and full length-level LOMETS threading to predict contact map and detect templates. FUpred and ThreaDom are used for domain boundary prediction based on the predicted contact map and threading templates, respectively. The individual domain-level sequences are put into the LOMETS threading algorithm again to generate the domain-level threading results (Figure 1, left). In domain-level LOMETS threading pipeline, MSA is used as inputs into 11 threading programs, which are all locally installed on our cluster, to identify structural templates from the PDB library. The MSA is also used to predict residue-residue contacts, distances, and hydrogen bond geometries, that are used in the 5 contact-based threading programs. These predicted terms along with the profile score from original profile-based threading are used to re-rank the templates detected by the individual threading programs. The top templates are ranked and selected by a score that combines the alignment Z-score, program-specific confidence scores and the sequence identity to the query. The functional annotations (including gene ontology terms, enzyme commission number, and ligand binding pockets) are generated by searching the template structures through the BioLiP function library [4]. Then, the 5 full-length models are constructed by L-BFGS system (DeepFold) using the distance restraints predicted by DeepPotential and calculated from top templates. FG-MD and FASPR will be used to refine the global topology and re-pack the side-chain conformation of the final domain models (Figure 1, right). Finally, the individual domain-level models and templates are then assembled into full-length models and templates by DEMO using the deep learning predicted distance restraints between domains, and structural analogs in PDB are detected by TM-align by matching the first LOMETS model to all structures in the PDB library. LOMETS reports the top 10 proteins from the PDB that have the closest structural similarity, i.e., the highest TM-score, to the predicted model, associated with the functional annotation. Furthermore, COFACTOR is modified by adding the LOMETS threading templates associated with structure models to predict protein functions, including Gene Ontology term (GO), Enzyme Commission number (EC), and Ligand Binding sites (LBS). A flowchart of the LOMETS pipeline is depicted in Figure 1, where users can find references for the individual threading methods at the bottom of the page.

Users can use LOMETS output to generate biological insights for their protein of interest. For example, the functional annotations of the targets given by LOMETS can tell users the type of enzymes the target proteins belong to (EC term), protein functions, such as protein-binding and ATP-binding, of the target proteins (GO term), and/or potential ligands and their respective binding site residues, so that users can reduce the scope of experiments based on the information.
For those users who want to quickly predict 3D models for a query sequence, detect its homologous templates and/or determine the functional annotations (GO terms, EC numbers, and ligand binding sites) for the detected templates, we recommend they use LOMETS. The LOMETS server does not attempt to extensively refine the threading models, so the response time is fast.
LOMETS is a meta-server method designed for protein structure prediction. It has two major advantages over other protein structure prediction servers. First, LOMETS can give users results quickly. Second, the quality of the structural models predicted by LOMETS are relatively high, even though they are slightly worse than I-TASSER.
Both LOMETS and I-TASSER are servers designed for protein structure prediction. Starting from a query sequence, the I-TASSER server first retrieves template proteins using LOMETS, and then performs structural refinement assembly simulations. Despite their accuracy, the refinement simulations are time-consuming. For those users who want a quicker response time or who do not need refined models, we recommend they use only LOMETS. Since the LOMETS server does not attempt to refine the threading models, the response time is faster than the I-TASSER server.
Second, since I-TASSER models are often structures combined from multiple templates, it is difficult or impossible to track the source of the original templates used to build the composite models. However, since LOMETS models are mostly derived from individual templates, the correspondence between the final models and the starting templates is more transparent. Partly due to the usefulness of template data transparency, LOMETS provides a longer list of template alignments (11*10=110 templates), while I-TASSER only lists the top-ten templates that are most influential to the final model construction.
Finally, both the LOMETS and I-TASSER servers give functional annotation information. But the functional annotations given by I-TASSER are predicted using our in-house COFACTOR server for query proteins. On the other hand, LOMETS shows the functional annotation information directly associated with the original homologous templates. Even so, since the query protein and the templates should be homologous, LOMETS can give users a general sense of the query function.
In summary, if users want to have a quicker response and pay more attention to the insights derived from the original homologous templates, we recommend they use LOMETS. However, if users want to construct high-quality model predictions of the 3D structure of a query protein, especially when the query protein may not have closely homologous templates, we recommend they use I-TASSER.
LOMETS has been updated to LOMETS3 with major updates, including:
For a given target, 220 templates are generated by 11 component servers, where each server generates 20 templates that are sorted by their Z-scores for each threading algorithm. The top 10 templates are finally selected from the 220 templates based on the following scoring function:
score(i,j)=conf(i)·Z(i,j)/Z0(i) + seqid(i,j)
where Z(i,j) is the Z-score of the j-th template for the i-th server, Z0(i) is the Z-score cutoff for defining good/bad templates for the i-th server, conf(i) is the confidence of the i-th server, which is defined as the average TM-score to the natives of all predictions calculated from a large-scale benchmark test. seqid(i,j) is the sequence identity to query for the j-th template of the i-th server. The parameters are listed in the following table:
i Server(i) Z0(i) conf(i) Reference
- --------- ------ ------- ---------
1 Hybrid-CEthreader 6.1 0.495 [5]
2 SparksX 7.8 0.478 [6]
3 CEthreader 6.0 0.472 [5]
4 HHsearch 22.0 0.471 [7]
5 MapAlign 3.8 0.471 [8]
6 MUSTER 8.5 0.461 [9]
7 MRFsearch 6.0 0.456 [10]
8 DisCovER 6.9 0.445 [11]
9 FFAS3D 46.0 0.440 [12]
10 EigenThreader 6.0 0.437 [13]
11 HHpred 83.0 0.389 [14]
The Z-score in the scoring function uses score terms from contacts, distances, and hydrogen bond geometries predicted by DeepPotential, and sequence profile score terms from the original profile-based threading methods:
Where \(Z\text{-}score^{MAE}(i,j)\) is the Z-score of the mean absolute error (MAE) based on predicted distance-map, \(Z\text{-}score^{CMO}(i,j)\) is the Z-score of numbers of overlapping contacts based on the predicted contact-map (CMO), \(Z\text{-}score^{HB}(i,j)\) is the Z-score based on the predicted hydrogen bond geometry (HB), and \(Z\text{-}score^{Prof}(i,j)\) is a score based on orignial profile threading scores.
The contact-map overlap is calculated from the number of overlapping contacts between the predicted contact-map and the contact-map derived from the aligned template, normalized by the number of contacts of the predicted contact-map.
The mean absolute error is calculated from the difference in the predicted query distance-map and distance-map derived from the aligned template.
The hydrogen bond score is calculated from the difference in hydrogen bond angles between the predicted query hydrogen angles and hydrogen angles derived from the aligned template.
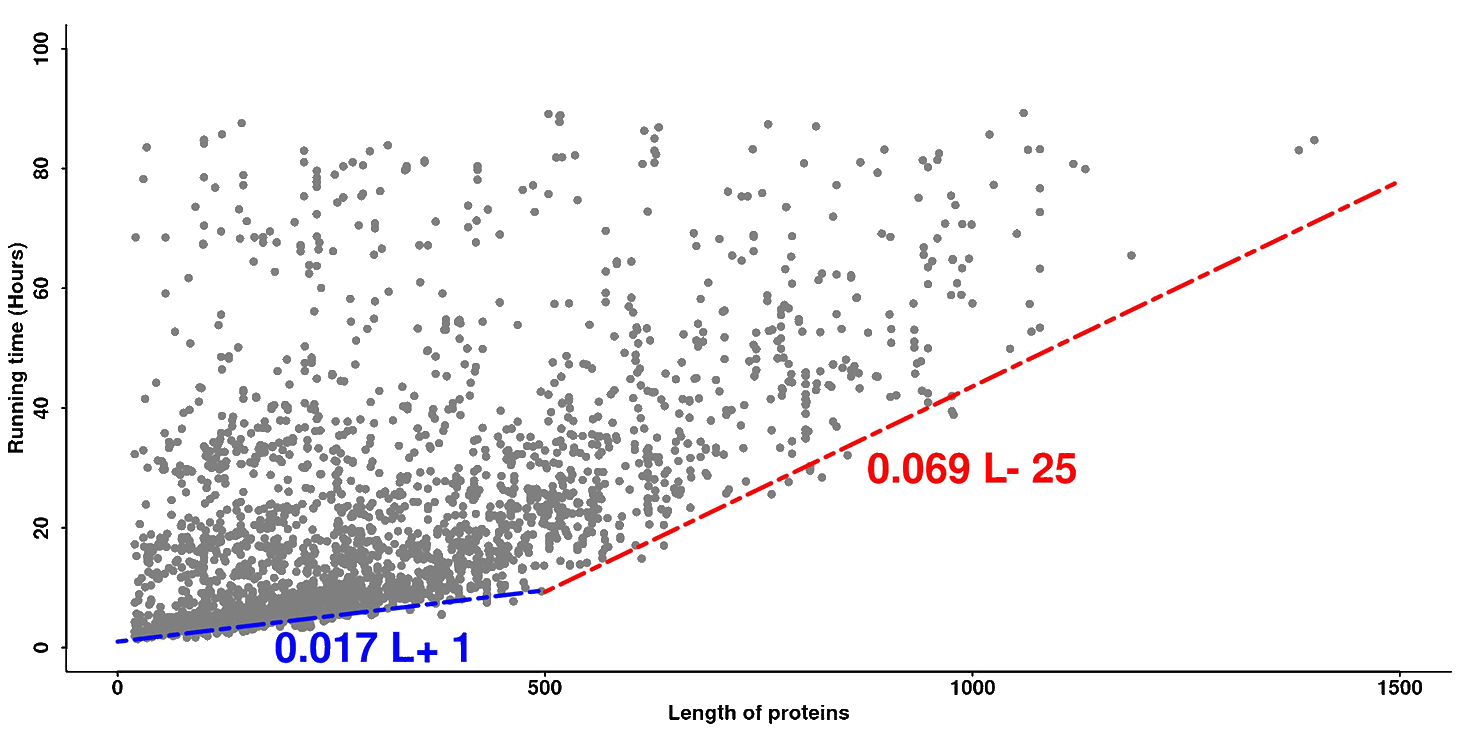
The running time depends on the protein size. Typically, a smaller protein takes less time than a larger protein. Furthermore, if too many sequences are accumulated in the queue, the procedure may take a longer time. Figure 3 represents the actual response time versus protein size for the 3,779 jobs processed by the LOMETS server recently. The blue line and red line are fit to the targets with the quickest response time, which should correspond to the actual running time of the LOMETS programs when the job queue is clear.
User Inputs
The user needs to paste the fasta-formatted amino acid sequence into the input box, or upload the amino acid sequence of the query protein using the browse button.
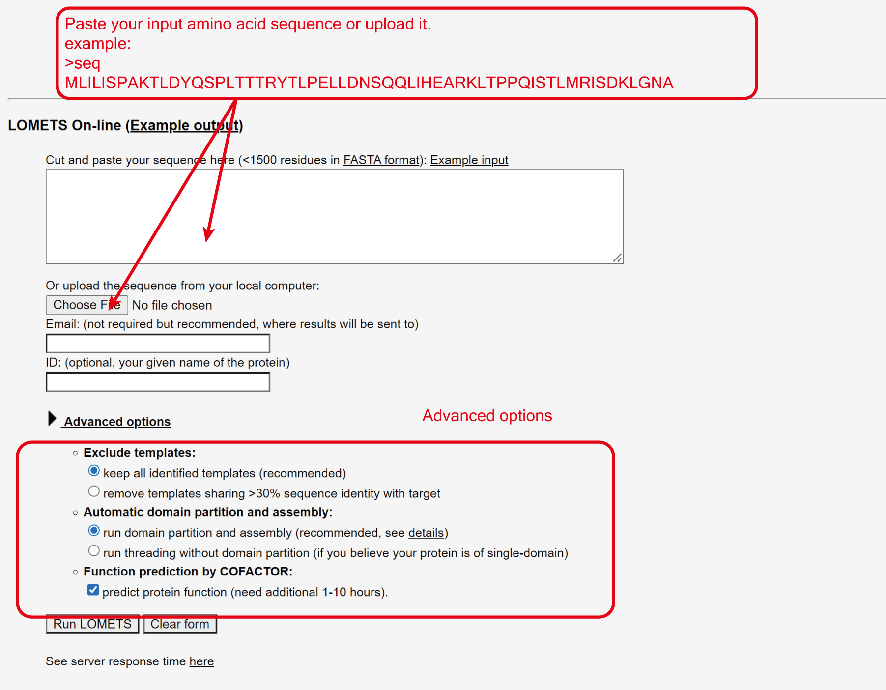
Figure 4. User inputs.
Advanced Options
Exclude templates: LOMETS derives models from known PDB structures (templates). If "remove templates sharing >30% sequence identity with target" is chosen, templates will not be generated from template structures that are highly homologous to the target sequence. In general, excluding homologous templates will make structure prediction harder, so this option is only for benchmarking purposes.
Automatic domain partition and assembly: LOMETS will automatically do domain partition for the query protein after the first round full length-level threading. Then assembly the domain-level templates and model to the full-length templates and models. Thus the running time will slightly longer than only run first round full length-level LOMETS threading. If you know the protein is a single-domain target or is a homologous target that has good templates can cover most of the sequence, you can select "run threading without domain partition" in "Advanced options" section below the input box, then the system will ignore the domain partition and assembly modules. Then LOMETS will only run one round full length-level threading which can make your job complete slightly faster.
Function prediction by COFACTOR: LOMETS will automatically do protein function prediction with a modified version COFACTOR. The function prediction step will take 1~10 hours after LOMETS model generated. If you do not need the function prediction and want to the job complete faster, you can un-check the box.
Content in output page
Illustration of output
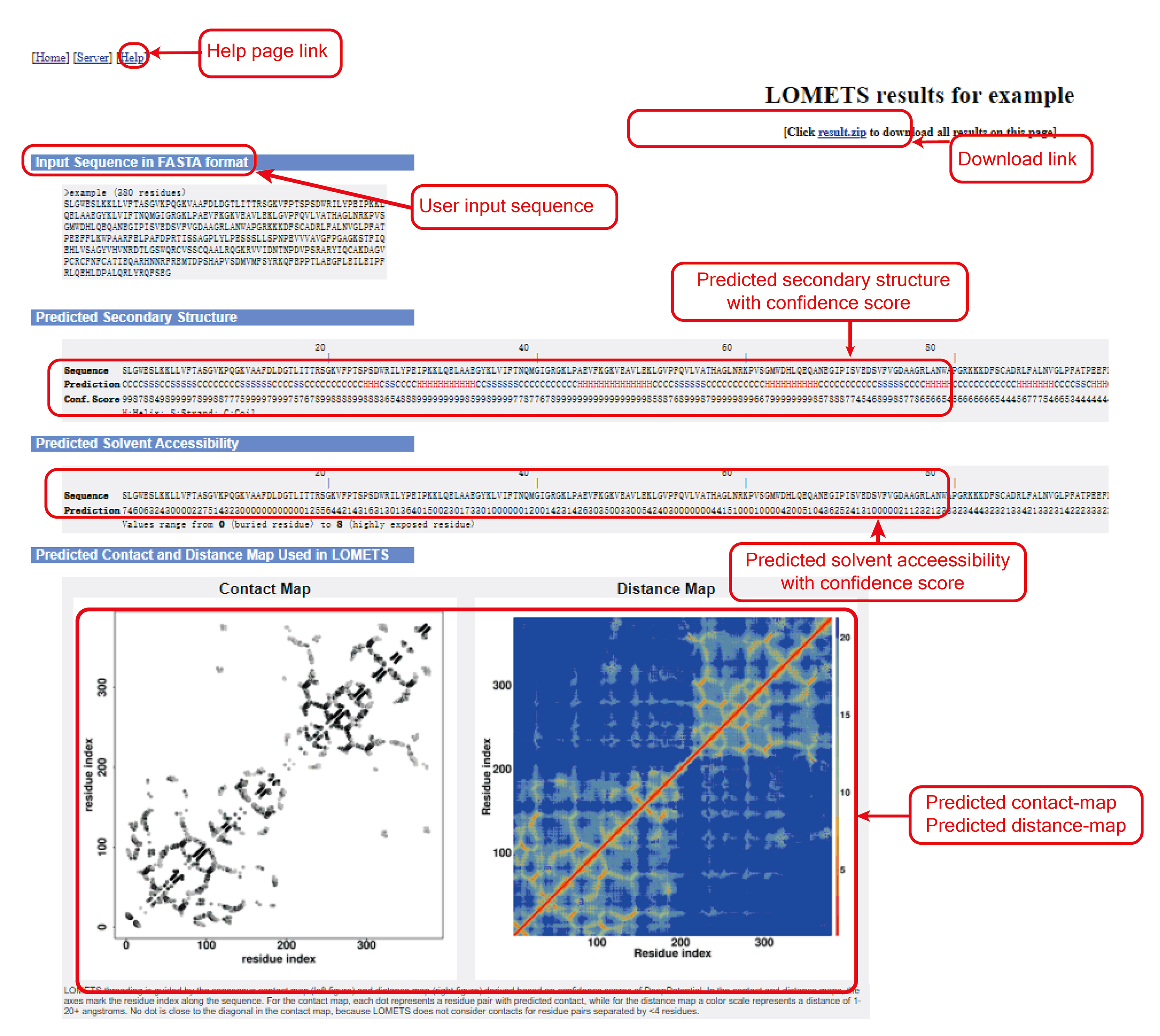
Figure 5. LOMETS summary output.
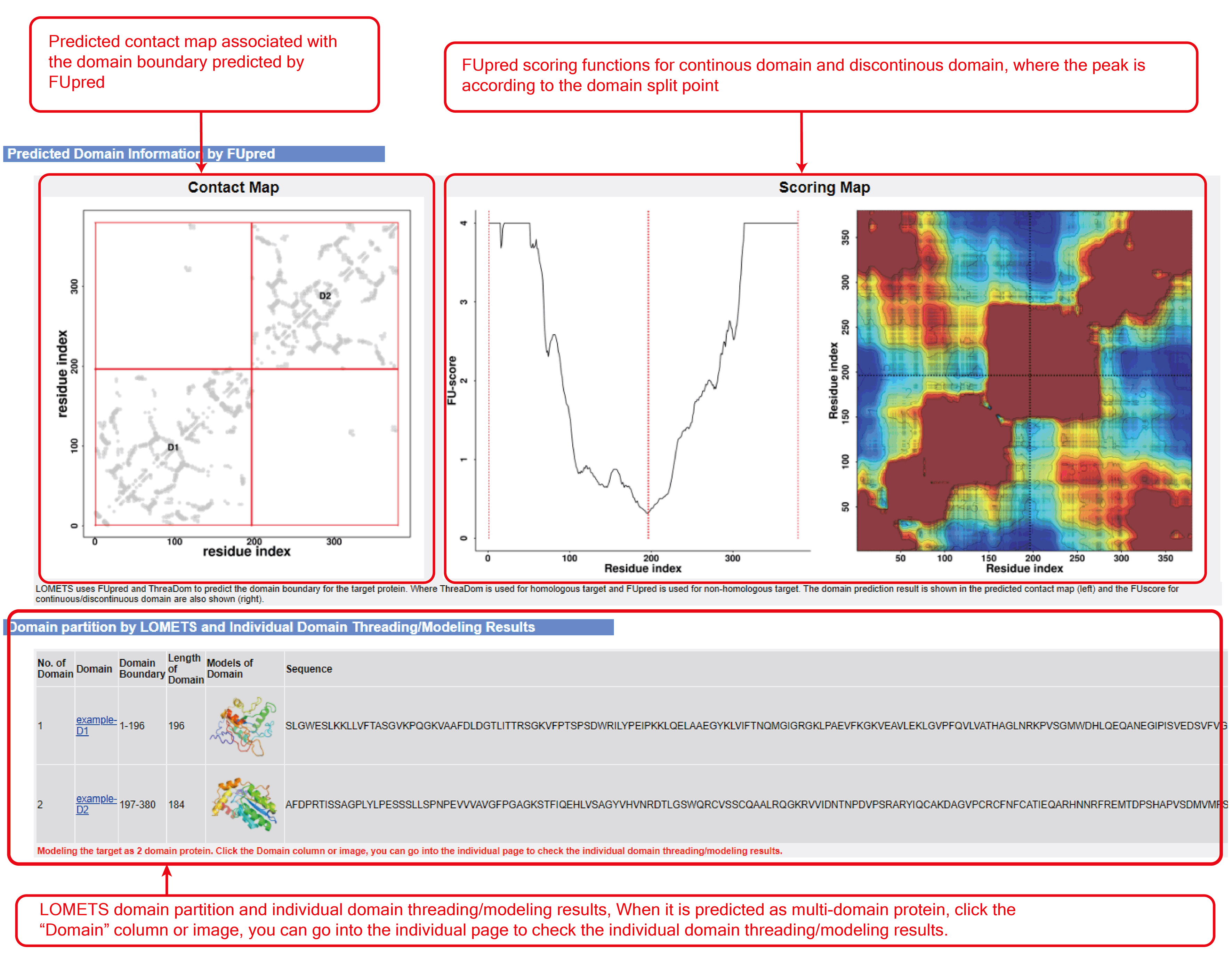
Figure 6. Domain partition and individual threading results (optional when "run domain partition and assembly" checked in "Advanced options" section).
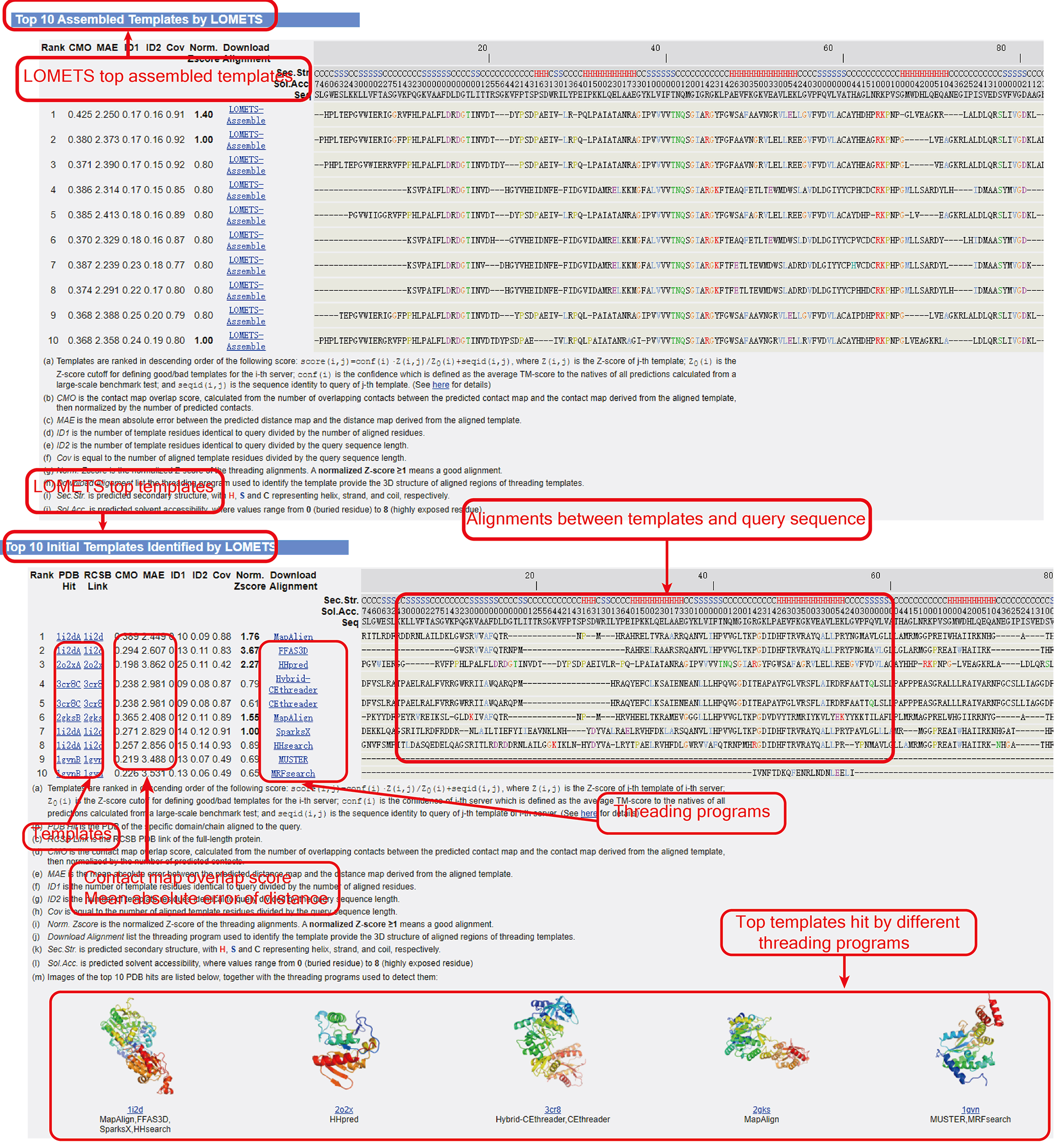
Figure 7. Top assembled templates (optional) and initial templates by LOMETS.

Figure 8. Models.

Figure 9. Structural analogs.
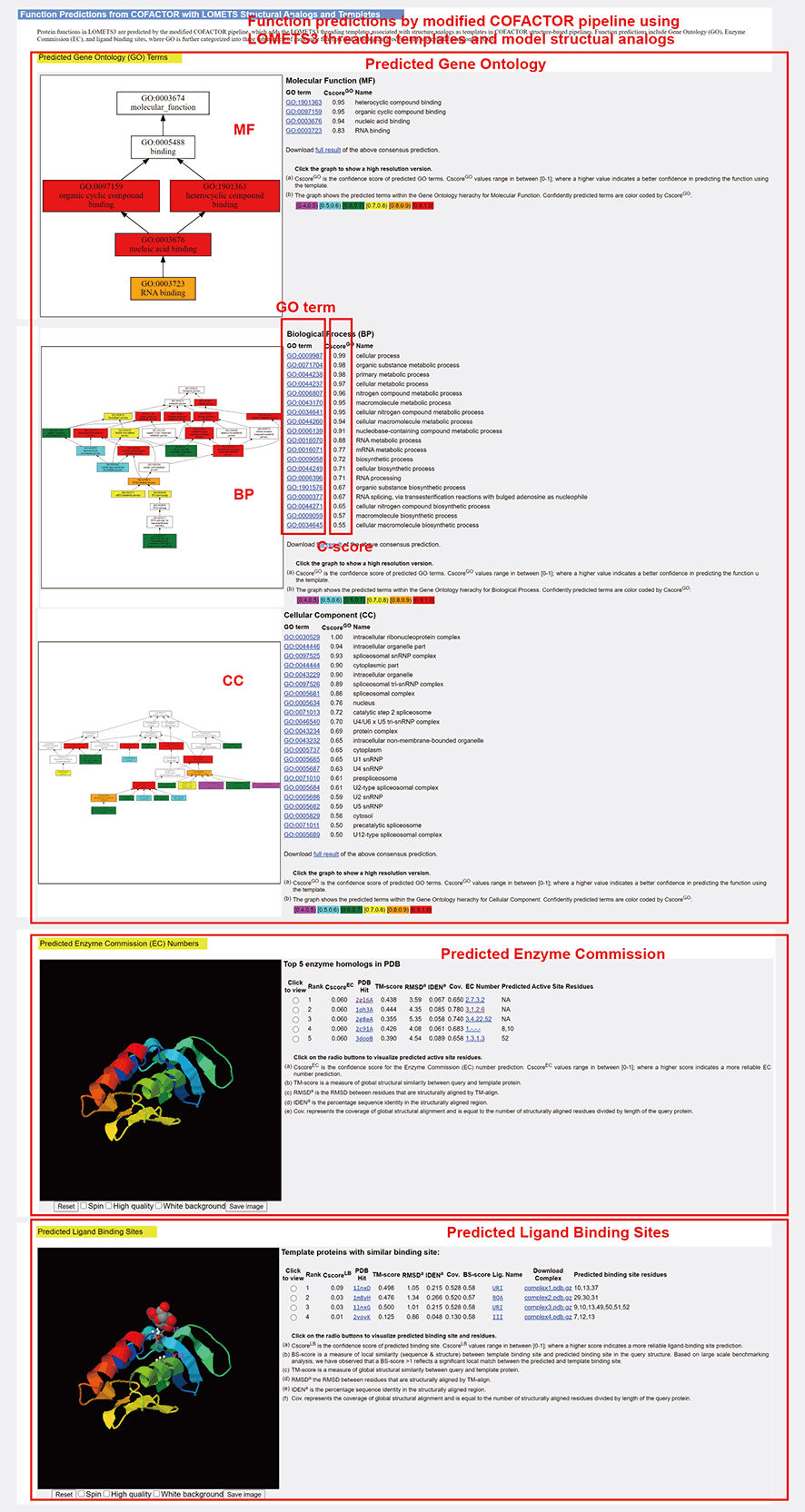
Figure 10. Function Predictions.

Figure 11. Individual threading results and functional annotations.
[1] Zheng, W., Wuyun, Q., Zhou, X., Li, Y., Freddolino, P., Zhang, Y. LOMETS3: Integrating deep-learning and profile-alignment for advanced protein template recognition and function annotation, in preparation, (2020).
[2] Zheng, W., Zhang, C., Wuyun, Q., Pearce, R., Li, Y., Zhang, Y. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Research, 47: W429-W436 (2019).
[3] Wu S, Zhang Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Research. 35, 3375-3382 (2007).
[4] Yang J, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions,
Nucleic Acids Research, 41: D1096-D1103 (2013)
[5] Zheng, W., Wuyun, Q., Zhang, Y. (2019) Detecting distant-homology protein structures by aligning deep neural-network based contact maps, PLOS Computational Biology, 15: e1007411.
[6] Zhou, H. and Zhou, Y. (2005) Fold recognition by combining sequence profiles derived from evolution and from depth-dependent
structural alignment of fragments. Proteins, 58, 321-328.
[7] Soding, J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics, 21, 951-960.
[8] Ovchinnikov S, Park H, Varghese N, Huang PS, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, Baker D. (2017) Protein structure determination using metagenome sequence data. Science, 355(6322):294-298.
[9] Wu S, Zhang Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information.
Proteins, 72, 547-556 (2008).
[10] Ma J, Wang S, Wang Z, Xu J (2014) MRFalign: Protein Homology Detection through Alignment of Markov Random Fields. PLOS Computational Biology 10(3)
[11] Bhattacharya S, Roche R, Bhattacharya D (2020)
DisCovER: distance-based covariational threading for weakly homologous proteins. Cold Spring Harbor Laboratory.
[12] Xu D, Jaroszewski L, Li Z, Godzik A. (2014) FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics. 30(5): 660-7.
[13] Buchan, D., & Jones, D. T. (2017). EigenTHREADER: analogous protein fold recognition by efficient contact map threading. Bioinformatics, 33(17), 2684–2690.
[14] Meier, A, and Söding J. Automatic prediction of protein 3D structures by probabilistic multi-template homology modeling. PLoS Comput Biol 11, no. 10 (2015): e1004343.
zhanglab![]() zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417
zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417